
上海纽约大学化学助理教授孙翔指导的研究团队提出了“分子动力学”领域的建模新方法,所建立的模型可计算电荷及能量在各类有机半导体材料中的转移速率与通道,并可用于评估量子动力学方法的有效性。这一研究成果发表于国际知名期刊《化学物理期刊》(The Journal of Chemical Physics),孙翔教授团队中的博士后胡竹斌和博士生Dominikus Brain分别为本研究论文的第一、第二作者。
“分子量子动力学”是理论与计算化学研究的核心主题之一,主要依靠计算机来模拟分子、原子体系的运动。“当原子和分子受到外界激发,例如被太阳光照射,其内部的电荷会产生正负分离,从而产生电流——这就是‘光诱导电荷转移’过程。我们可以模拟这一过程,测算凝聚态环境中电荷转移的速率和通道,从而为太阳能转化效能的研究奠定理论基础,有助于设计发明更高效的能量转化材料。”孙教授介绍说。
分子有不同的电子态。“例如,当分子中的电子都处在最低能量下,这就是‘基态’;当电子受到光照刺激‘兴奋’起来,这时候的状态就是‘激发态’。”孙教授说。在该领域的研究中,针对这两个电子态的光诱导电荷转移过程,已建起成熟的谐振模型;但尚未开发出针对多个电子态的有效建模方法。
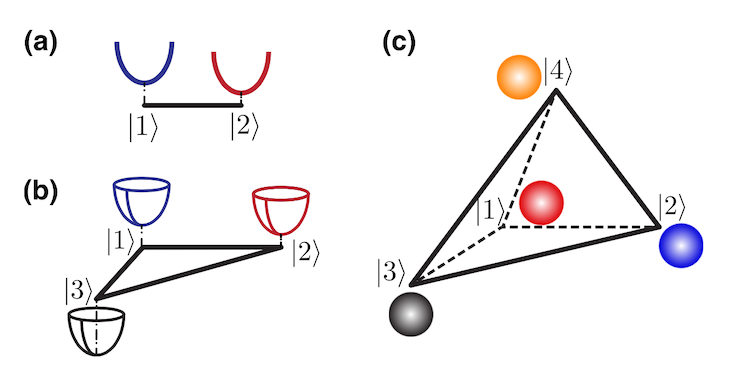
势能面图示:(a)两态模型;(b)三态模型;(c)四态模型
孙翔教授团队在本次研究中,通过完成大量理论计算,成功提出构建多态谐振模型的新方法,可在任意数量电子态的情况下模拟光诱导电荷转移的过程。研究中的计算资源支持来自上海纽约大学超算平台。“新模型为在复杂体系中进行非绝热动力学的模拟提供了通用且灵活的框架。更重要的是,这套模型体系可作为评估标准,检验其他动力学方法的有效性,并可以就不同材料的研究选择恰当的研究方法。”孙教授说。
计算化学是上海纽约大学重点发展的研究方向之一。“‘小而精’是我们的团队特色。也正得益于团队不到10人的规模,我们得以针对每位同学的具体情况和需求开展个性化的指导。华东师范大学-纽约大学计算化学联合研究中心(上海纽约大学)借助其独特的定位,发挥纽约大学全球教育体系的平台优势,与国内外重点大学及前沿科研机构之间联系紧密,促进中心与其他学术机构开展密切的沟通与合作,使得我们能够紧随国际上最前沿的研究趋势与动态。”孙教授说。

孙翔教授团队合影
论文第一作者胡竹斌完成在上海纽约大学的博士后研究工作后,现已加入华东师范大学担任专职副研究员。第二作者Dominikus Brain则将继续在孙翔教授团队完成博士阶段的学习和研究。本次研究获得了国家自然科学基金的支持。